CSC277 - Project 1
Simulating the Cell
Overview
For this project, we will explore object-oriented programming by creating three classes, DNA, RNA
and Protein.
Description
 DNA is composed of
long strands of four basic molecules: Adenine, Cytosine, Guanine and Thymine. These four bases coil
together to form a double-helix structure, and we have 23 such structures, called chromosomes, in
every cell of our body. The bases have the interesting property of pairing up across the
double-helix, such that Adenine always matches Thymine, and Cytosine always matches Guanine.
For our purposes as computer scientists, we can abbreviate these bases as A, C, G, and T.
DNA is composed of
long strands of four basic molecules: Adenine, Cytosine, Guanine and Thymine. These four bases coil
together to form a double-helix structure, and we have 23 such structures, called chromosomes, in
every cell of our body. The bases have the interesting property of pairing up across the
double-helix, such that Adenine always matches Thymine, and Cytosine always matches Guanine.
For our purposes as computer scientists, we can abbreviate these bases as A, C, G, and T.
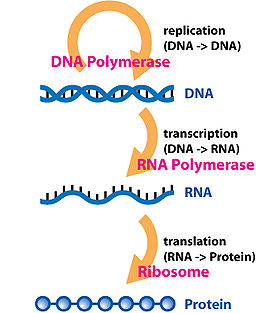
DNA contains segments called genes detailing how our cells should work.
A gene becomes a protein through a process known as the
Central dogma
of molecular biology: DNA is transcribed into RNA (Ribonucleic Acid) and then
translated
into proteins. Proteins are the vital machinery that makes our cells work; they form the cell walls
and transport molecules around the cell. It is this process
of DNA to RNA to Proteins that we will simulate in this project by creating
appropriate classes and objects.
Because of how genes are encoded, most
genes have a balanced number of Gs and Cs in relation to the number of As and Ts. To determine the
G-C content of a DNA strand, we can sum the number of Gs and Cs we find, and divide this by
the total number of bases in the string. Most genes have between 25% and 75% Gs and Cs.
Our next step is to transcribe the DNA into RNA. RNA is a single-stranded molecule as opposed to
the double-stranded DNA. This means it cannot replicate itself as does DNA, but RNA still
plays many useful roles in cell function. Primarily, RNA gets translated into
proteins. Having these steps be separate eases the burden on DNA to process all the work.
In the transcription
from DNA to RNA, every Thymine molecule is replace with Uracil (denoted with U).
When you transcribing DNA into a strand of RNA, there are two important locations to find: where to
start
reading and where to stop reading.
This process is rather complex, and so we will simplify here to only transcribe the exact location
of a gene (in reality there are buffers transcribed before and after the entire gene).
Just like words are composed of individual letters, genes are divided up into segments called
codons. A codon consists of three sequential bases, so with 4 choices for each base
(from A, C, G, and T), this gives us 4 X 4 X 4 = 64 different combinations.
So there are actually three different ways to change a strand into amino acids.
We could start with the first base at index 0,
such that our codons are substrings from 0:3, 3:6, 6:9, etc. Staring with index 1
makes our substrings 1:4, 4:7, 7:10, etc, and starting with index 2 gives us 2:5, 5:8, 8:11, etc.
These initial indices (0, 1, 2) define the reading frame for this gene.
For example, in English we start every sentence with a capital letter, and stop each sentence with a period.
DNA uses an interesting way of signaling the start of a gene by designating one particular codon,
ATG, as the start codon. For our purposes, wherever you find ATG, this marks the start of a gene;
this also defines our reading frame, which can be found by modding the initial index of the start
codon by 3.
For robustness, there are three codons which tell us the end of a gene: TAG, TGA and TAA.
As soon as any one of these three stop codons is found
in the same reading frame after the start codon,
we have found the end of the gene and our RNA molecule.
With our strand now in RNA form, we are ready to make the final step to proteins. This works
by reading the Genetic Code of RNA.
Each of the 64 possibile codons translates into one of 20
amino acids
(redundancy is built into this system to decrease the effects of random mutations),
and it is the conjunction of these amino acids that ultimately fold up and create proteins.
This can be represented in a
Codon Table, pairing codons such as UCU, UCC, UCA and UCG with
Serine. See the paragraph before the Testing section for more
details on how to read this Codon Table.
(Note, the amino acids are commonly abbreviated to a single letter, like DNA molecules,
so in the case of Serine, this would be S.)
In this project, you will simulate this process as described above using object-oriented
programming in Python to create three classes, DNA, RNA and Protein, along with a driver program
for the cell.
Coding
You will be writing two files for this project.
pgsm.py
First, pgsm.py (which stands
for Protein, Gene and Small Molecule) will contain your
class definitions for DNA, RNA and Protein. Each of these classes is described below.
DNA
The DNA class will represent a chromosome within a cell. It will bring in a string of DNA
and be able to return a list of all genes contained within this DNA string or its reverse
complement.
__init__(self, strand)
The constructor brings in a string called strand, which will be saved as a data member component
of this DNA object.
__str__(self)
This method will be called by str() to produce a string representing the DNA object. If the strand is longer than 33
bases, return the first 15 bases, followed by "..." and then the last 15 bases, otherwise,
return the whole DNA strand.
length(self)
This method will return the length of the strand for this DNA object.
gc_content(self)
Calculate the G-C content of the DNA object and return as a floating point number. This
will represent the percent of Gs and Cs in the strand as compared to the complete length of
the strand. You should implement the search for Gs and Cs yourself such that you run through the
strand only once, as opposed to using the find method of string objects.
For half credit, you can use the count method.
invert(self)
DNA is a double-stranded molecule. The strand passed in through the constructor is only one half
of the molecule. This method will replace that strand with it's reverse complement. You can find
the opposite strand of DNA by replacing all Gs with Cs and vice versa, and all As with Ts and
vice versa, and then reversing the string. Using a recursive definition to reverse the string
will be infeasible when handling 1 million base-pair DNA molecules; instead, you should use
a for-loop for this reversal.
transcribe_genes(self)
This method returns a list of RNA objects representing transcribed genes based on current the
inversion of the DNA strand. To find the RNA objects, walk through the string searching for the
start codon "ATG" and record these indicies in a list.
Then for each start codon, search the rest of the string to find a stop codon "TAG", "TGA" or "TAA"
in the same reading frame. Your RNA strands will be the base pairs between each start and stop
codon; use these to create RNA objects to add to your list. You should substitute a U for each T
found in this substring before creating the RNA object.
It is possible that genese will overlap, containing some of the same
DNA but in different reading frames. If you reach the end of a DNA strand before you find a Stop
codon in the same reading frame, do not create an RNA molecule.
RNA
__init__(self, r_strand, start, stop)
The constructor for an RNA object will bring in a string called r_strand, along with the
start and stop index of transcription from the original DNA molecule. All of these will
be saved as a data member components of this RNA object.
__str__(self)
This method will be called by str() to produce a string representing the RNA object.
First, the string will contain the start and stop indicies of the RNA, separated by a dash.
If the strand is longer than 33
bases, return the first 15 bases, followed by "..." and then the last 15 bases, otherwise,
return the whole RNA strand.
translate(self, codon_table)
This method will return a Protein object based on the RNA strand and the dictionary
codon_table. For each three letter codon, translate it to the appropriate
amino acid based on the codon_table. Create a Protein object with this
amino acid string and the start and stop index, and return it.
Protein
__init__(self, aa, start, stop)
The constructor for a Protein object will bring in a string called aa, along with the
start and stop index of transcription from the original DNA molecule. All of these will
be saved as a data member components of this Protein object.
__str__(self)
This method will be called by str() to produce a string representing the Protein object.
First, the string will contain the start and stop indicies of the Protein, separated by a dash.
If the strand is longer than 33
amino acids, return the first 15 amino acids, followed by "..." and then the last 15
amino acids, otherwise,
return the whole Protein strand.
cell.py
Second, cell.py will contain the main function for the program. You will implement
the following pseudocode:
- Ask the user what file of DNA they wish to analyze and load this file
- Ask the user what file contains the Codon Table and load this file into a dictionary
- Create a DNA object based on the data contained in the DNA file
- Print the string representation, length, and G-C content of the DNA object
- Transcribe the DNA to get a list of RNA molecules
- Print the number of RNA molecules found
- Open a file with the same name as the DNA file, but with ".pro" attached to the end
- For each RNA molecule r
- Translate r into a Protein
- Print the Protein to the screen
- Write the Protein as a line to the file with ".pro" attached to the end.
- Close the protein file.
The DNA file will be in a format such that the data might be split over multiple lines.
The RNA Codon Table file is organized to have the letter of an amino acid first, followed by
the codons which encode for this amino acid separated by commas.
The Codon table should be loaded such that the keys are 3 base pairs of RNA (such as GCU, AUU) and
the values are the single letters representing an amino acid. Note that not all methods
requested above, such as invert, are used in cell.py. These methods
will be tested separately during grading.
Testing
Download the small DNA file to test your program:
This is one DNA molecule, but it is split over more than one line. It includes
DNA which encodeds more than one protein. Use this file to test and debug your code above.
Your output should be as follows:
DNA Molecule is TTAATAGCGTGGAAT...GTCCTAAAGATAACA
Length is 99 base pairs
GC Content is 0.42424242424242425
Found 2 RNA molecules
The Proteins are:
1: 16-48 ILIKECHEESE
2: 58-87 EATVEGGIES
Next, download the file:
It contains the complete genome for a species of bacteria, which is stored as
one large circular DNA strand. Use the first 500 lines of the
file to test your cell.py program.
According to
current
analysis, E. coli UTI89 should contain
approximately 5000 genes total, many less than what would be predicted by our program.
The discrepancy is due
to the simplifications we made in our model of DNA; in reality, there are many other factors
beyond the start codon which determine if a segment of DNA encodes a gene, such as
Transcription Factors and
Promoters.
You must hand in:
- cell.py
- pgsm.py
- ecoli_uti89.pro
and any other files necessary to run your code.
© Mark Goadrich 2010, Centenary College of Louisiana